Decoding the Tumor Microenvironment: A Deep Dive into Spatial Proteomics,Transcriptomics,and the Microbiome
Understanding the complex interplay within the tumor microenvironment is crucial for developing effective cancer therapies. Recent advancements in spatial technologies allow us to move beyond bulk analysis and examine these interactions with unprecedented detail. This article details the analytical approaches used in a recent study investigating the relationship between the intratumoral microbiome, protein expression, and gene transcription in glioma tumors. We’ll break down the methodologies employed, offering clarity for researchers and those interested in this rapidly evolving field.
Data Acquisition and Initial Processing
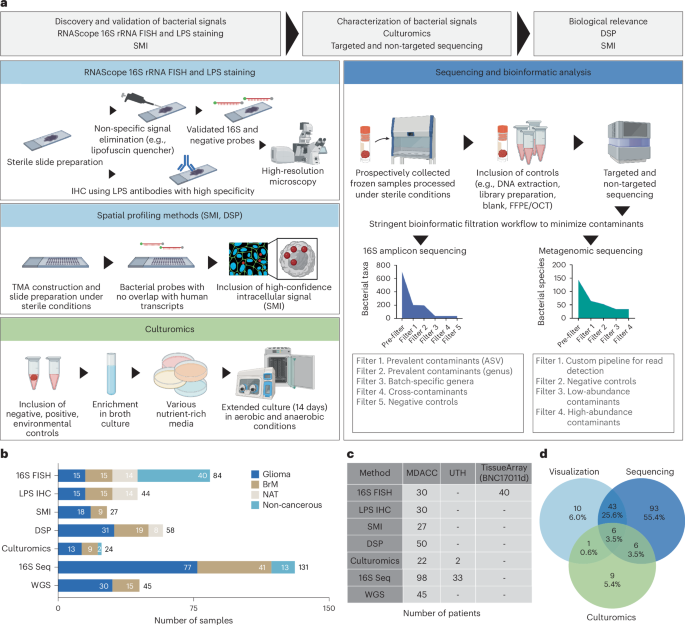
The foundation of this research involved a multi-omic approach, integrating data from 16S rRNA gene sequencing (to characterize the microbiome), spatial transcriptomics, and spatial proteomics. Here’s a look at how the data was prepared for analysis:
* Proteomic Data: Raw protein signals were initially processed by thresholding, ensuring only reliable data points were considered. Background normalization, using immunoglobulin Gs (IgGs), was then applied to refine the signal and reduce noise.
* Transcriptomic Data: RNA data underwent log2 normalization to stabilize variance and facilitate comparisons.
* Microbiome Data: 16S rRNA gene sequencing data was used to categorize regions as either “16S-high” or “16S-low” based on bacterial signal intensity.
Unraveling Data Complexity: Statistical Approaches
To make sense of the vast datasets generated,a suite of refined statistical methods were employed. These techniques aimed to identify key differences between 16S-high and 16S-low regions, and to correlate these differences with clinical outcomes.
1.Principal Component analysis (PCA) & PERMANOVA:
PCA was used to visualize the overall data structure and identify major patterns. Specifically, log2-normalized transcriptomic data was subjected to PCA with centering and scaling. PERMANOVA (Permutational Multivariate Analysis of Variance) – using the adonis function in the vegan R package – then assessed whether the observed differences between 16S-high and 16S-low regions were statistically critically important.This helps determine if the microbiome’s presence fundamentally alters the overall molecular landscape.
2. Linear Mixed models (LMM): Identifying Differential Abundance
LMMs were the workhorse for identifying proteins and transcripts that differed considerably between 16S-high and 16S-low regions. Here’s how it worked:
* Accounting for Biological Variation: LMMs were used to account for patient-specific variability by including patient ID as a random intercept. This is crucial becuase each patient’s tumor is unique.
* Controlling for Confounding factors: In the glioma analysis, the model was further adjusted to control for IDH status and tumor recurrence, providing a more accurate assessment of the microbiome’s impact. Two ROIs from one patient lacking these annotations were excluded from this specific analysis.
* Multiple Hypothesis Correction: The Benjamini-Hochberg procedure was applied to control for the increased risk of false positives when testing thousands of genes and proteins simultaneously.
* Importance Threshold: proteins and transcripts with an adjusted P* value < 0.05 and a log2 fold change (FC) > 0.58 were considered significantly different. These findings are detailed in Supplementary Table 5.
3. Pathway and Gene Enrichment Analysis
To understand the *functional consequences of differential gene and protein expression, pathway enrichment analysis was performed. This involved:
* Generalized LMM (SMI): Used for spatial metabolomics data.
* LMM (DSP & SMI): Employed for spatial proteomics and transcriptomics pathway analysis.
* Multiple Hypothesis Correction: Again, the Benjamini-Hochberg procedure was used to ensure robust results.
* Significance Threshold: Adjusted *P* values < 0.05 were considered significant.
4. Microbiome Data Analysis
Differential abundance of microbial taxa was assessed using two leading microbiome analysis tools:
* MaAsLin2 (version 1.20)
* ANCOM-BC (version 2.8.1)
these tools are designed to handle the complexities of microbiome data, including compositional effects
Related reading