
When the brain begins to resemble a sponge, riddled with microscopic holes where healthy tissue once thrived, the diagnosis is often grim: a prion disease. These rare, relentless neurodegenerative disorders — including Creutzfeldt-Jakob disease (CJD), variant CJD, and fatal familial insomnia — are caused not by viruses or bacteria, but by misfolded proteins that trigger a catastrophic chain reaction in the brain. Once considered medical curiosities, prion diseases have gained renewed attention due to their rapid progression, diagnostic challenges, and the eerie way they erase personality, memory, and motor function within months.
Unlike infectious agents that contain genetic material, prions are abnormal versions of a naturally occurring protein called prion protein (PrP). When these malformed proteins encounter their healthy counterparts, they induce them to misfold as well, setting off a domino effect that destroys neurons and leaves behind sponge-like vacuoles in brain tissue — hence the term “spongiform encephalopathy.” This process is not only swift but largely irreversible, with most patients deteriorating rapidly after symptom onset.
According to the World Health Organization, sporadic Creutzfeldt-Jakob disease — the most common form, accounting for about 85% of cases — occurs spontaneously in roughly one to two people per million annually worldwide. While the exact trigger remains unknown, it is believed to arise from a random conformational change in the prion protein. Inherited forms, linked to mutations in the PRNP gene, make up 10–15% of cases, while acquired forms — such as variant CJD tied to bovine spongiform encephalopathy (BSE), or “mad cow disease” — represent less than 1% but have drawn significant public health concern due to their potential for iatrogenic or dietary transmission.
The speed of progression is one of the most alarming features of prion diseases. As noted in clinical studies, patients often go from subtle cognitive changes to severe disability and death within six months. Early symptoms may include memory lapses, behavioral changes, or visual disturbances — signs easily mistaken for stress, depression, or more common dementias like Alzheimer’s. But, unlike Alzheimer’s, which typically unfolds over years, prion diseases accelerate with shocking velocity, leaving families and clinicians little time to intervene.
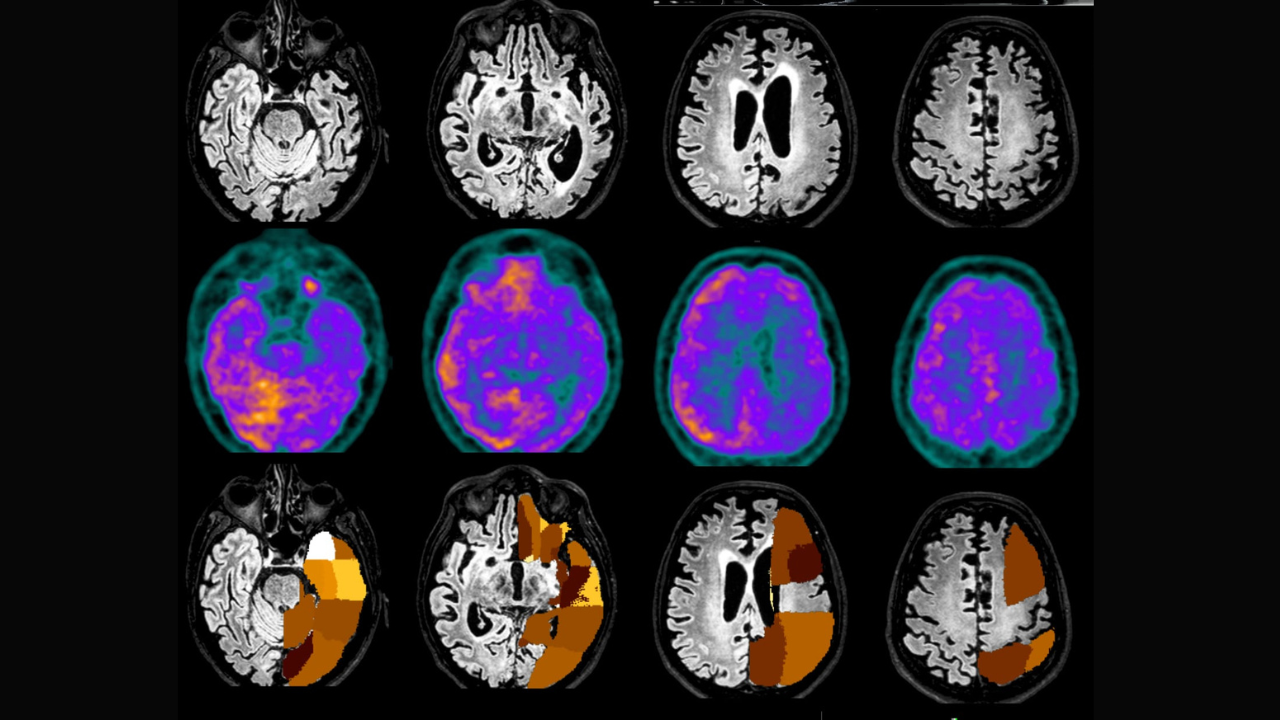
Diagnosis remains a significant hurdle. There is no single blood test or routine scan that can confirm prion disease definitively during life. Clinicians rely on a combination of neurological exams, electroencephalograms (EEGs) showing characteristic periodic sharp wave complexes, magnetic resonance imaging (MRI) revealing cortical ribboning or basal ganglia hyperintensity, and cerebrospinal fluid analysis for elevated biomarkers like real-time quaking-induced conversion (RT-QuIC), which has improved sensitivity in recent years. A definitive diagnosis, however, still requires neuropathological examination of brain tissue after death.
There are currently no curative treatments for any form of prion disease. Management focuses on symptomatic relief — addressing agitation, pain, and myoclonus (sudden muscle jerks) — and supportive care. Experimental therapies, including monoclonal antibodies and antisense oligonucleotides targeting the PRNP gene, are under investigation in preclinical and early clinical trials, but none have yet demonstrated efficacy in halting or reversing disease progression in humans.
Public health vigilance remains critical, particularly regarding iatrogenic transmission. Cases have been linked to contaminated neurosurgical instruments, corneal transplants, and pituitary-derived hormone treatments administered decades ago. Strict sterilization protocols — now including extended autoclaving and employ of disposable tools for high-risk procedures — have significantly reduced such risks. Similarly, rigorous surveillance of bovine populations and bans on specified risk materials in animal feed have helped prevent new variant CJD cases linked to BSE exposure.
Research into prion biology continues to yield insights relevant beyond these rare disorders. The prion-like mechanism of protein misfolding and propagation has been implicated in more common neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and amyotrophic lateral sclerosis (ALS), where abnormal aggregates of tau, alpha-synuclein, and TDP-43 spread through neural networks in a prion-like fashion. Understanding how prions propagate may therefore inform broader strategies against neurodegeneration.
For families affected, the emotional toll is profound. The rapid loss of a loved one — often a cognitively vibrant adult — to a disease that progresses with such ferocity can feel surreal. Support organizations, including the Creutzfeldt-Jakob Disease Foundation and national CJD surveillance units, offer resources, counseling, and registries to aid in both patient care and epidemiological tracking.
As surveillance improves and diagnostic tools like RT-QuIC become more widely available, experts hope to identify cases earlier, even if treatment remains elusive. Meanwhile, ongoing research into gene-silencing approaches and immune-based interventions offers cautious optimism for future breakthroughs.
The next scheduled update on global prion disease surveillance is expected from the World Health Organization in late 2024, following its biennial review of zoonotic and rare disease trends. For now, awareness, early recognition of red-flag symptoms, and adherence to infection control safeguards remain the best tools available.
If you or someone you know is experiencing rapidly progressive neurological symptoms, seek evaluation from a neurologist or cognitive disorder specialist promptly. Share this information to help others recognize the signs — and stay informed through trusted health authorities.