
The U.S. Food and Drug Administration (FDA) has granted accelerated approval to Otarmeni (lunsotogene parvec-cwha), marking the first-ever gene therapy approved for the treatment of genetic hearing loss. This landmark decision, announced on April 23, 2026, represents a significant advancement in addressing OTOF-related deafness, a condition caused by biallelic variants in the OTOF gene that disrupts sound signal transmission due to the absence of otoferlin protein.
Otarmeni is indicated for pediatric and adult patients with severe-to-profound and profound sensorineural hearing loss (any frequency >90 dB HL) associated with molecularly confirmed biallelic variants in the OTOF gene, provided they have preserved outer hair cell function and no prior cochlear implant in the same ear. Prior to this approval, no disease-modifying treatments existed for OTOF-related deafness, which accounts for approximately 2% to 8% of inherited, non-syndromic cases of congenital hearing loss.
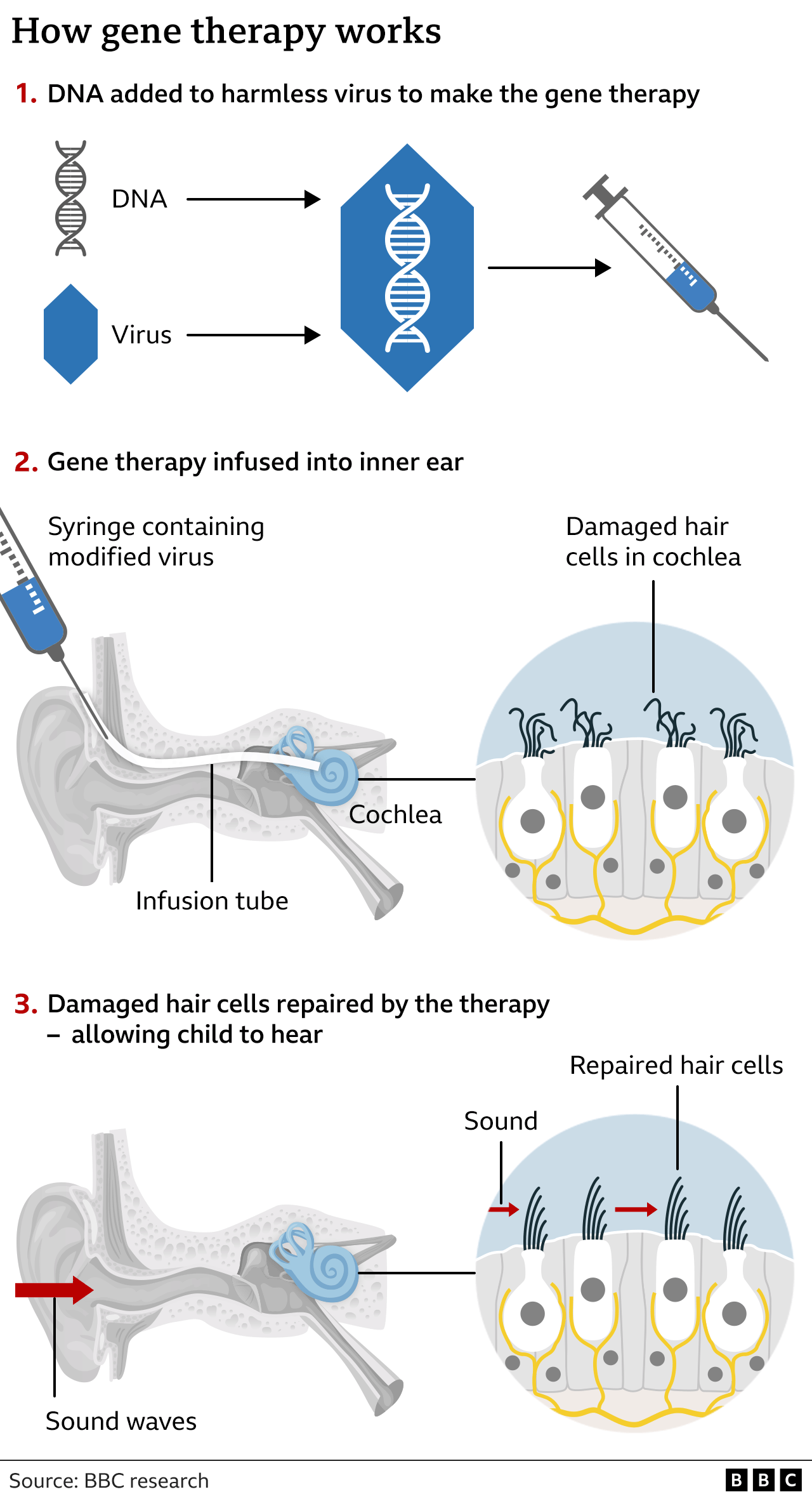
The therapy utilizes a dual adeno-associated virus (AAV) vector-based approach to deliver functional copies of the OTOF gene to the inner ear. This innovative delivery system aims to restore otoferlin production, thereby enabling proper sound signal transmission from hair cells to the auditory nerve. The FDA’s approval follows the publication of clinical trial results in the New England Journal of Medicine, which demonstrated meaningful hearing restoration in treated patients.
FDA Commissioner Marty Makary, M.D., M.P.H., highlighted the significance of the approval, stating that it underscores the agency’s commitment to accelerating therapies for rare diseases with unmet medical needs through the National Priority Voucher (CNPV) pilot program. Otarmeni is the sixth approval under this program and the first gene therapy product to receive authorization via this pathway. The Biologics License Application (BLA) for Otarmeni was approved in just 61 days after filing, tying it for the fastest BLA approval in modern FDA history.
The accelerated approval was granted based on surrogate endpoints reasonably likely to predict clinical benefit, with the requirement for a post-marketing confirmatory trial to verify and describe the therapy’s clinical benefit. Regeneron, the developer of Otarmeni, has committed to providing the treatment to eligible patients and conducting the necessary follow-up studies to support potential conversion to traditional approval.
Genetic mutations are responsible for about half of all congenital hearing loss cases. Variants in the OTOF gene are among the most common genetic causes of non-syndromic hearing loss, particularly in populations with high rates of consanguinity. Children with OTOF-related deafness often present with normal auditory brainstem responses but absent otoacoustic emissions, a pattern known as auditory neuropathy spectrum disorder. Without intervention, delayed diagnosis can lead to missed treatment windows and lasting speech and language developmental delays.
How Otarmeni Works: Mechanism of Action
Otarmeni employs two separate AAV vectors to overcome the packaging limitations of single-vector systems, as the OTOF gene complementarily exceeds the capacity of a single AAV capsid. One vector delivers the 5’ portion of the OTOF gene, whereas the other delivers the 3’ portion. Once inside the target hair cells, the homologous sequences recombine to form a full-length, functional OTOF transcript. This dual-vector strategy enables the expression of otoferlin, a calcium-sensitive protein essential for vesicle fusion and neurotransmitter release at the ribbon synapse of inner hair cells.

By restoring otoferlin function, Otarmeni aims to re-establish the synaptic transmission of sound signals from the inner hair cells to the auditory nerve. This mechanism addresses the root cause of OTOF-related deafness rather than merely amplifying sound, as hearing aids or cochlear implants do. The therapy is administered via a single intracochlear injection, targeting the scala tympani of the cochlea.
Clinical Evidence and Trial Results
The FDA’s decision was primarily informed by data from a Phase 1/2 clinical trial evaluating the safety and efficacy of Otarmeni in pediatric and adult patients with biallelic OTOF mutations. Key endpoints included changes in auditory brainstem response (ABR) thresholds, otoacoustic emissions, and speech perception measures. Preliminary results showed statistically significant improvements in hearing thresholds across multiple frequencies, with some patients achieving thresholds within the mild to moderate hearing loss range.
Adverse events observed in the trial were generally mild to moderate and included transient vestibular symptoms, procedural discomfort, and temporary elevations in liver enzymes. No serious adverse events related to the gene therapy vector were reported during the follow-up period. Long-term safety monitoring remains ongoing as part of the post-marketing requirements.
The trial included participants aged six months and older, with stratification by age and baseline hearing function. Subgroup analyses suggested that younger patients, particularly those under five years of age, exhibited more robust responses, likely due to greater neural plasticity and reduced cumulative auditory deprivation. These findings support early intervention as a critical factor in maximizing outcomes.
Impact on Patients and Families
For families affected by OTOF-related hearing loss, the approval of Otarmeni offers a potential disease-modifying alternative to conventional management strategies. While cochlear implants remain highly effective for many individuals with severe-to-profound hearing loss, they require surgical implantation, ongoing device maintenance, and do not restore natural biological hearing. A successful gene therapy could provide a more physiologic hearing experience, potentially improving music perception, sound localization, and speech understanding in noisy environments.
Early access to Otarmeni may also reduce the cumulative burden of auditory deprivation during critical periods of language development. Children who receive timely intervention may experience improved speech and language acquisition, reduced need for intensive speech therapy, and better educational and social outcomes. However, access to the therapy will depend on factors such as insurance coverage, geographic availability of specialized treatment centers, and eligibility criteria based on genetic confirmation and cochlear status.
Genetic testing for OTOF variants is recommended for infants who fail newborn hearing screening and demonstrate the auditory neuropathy profile (present ABR with absent OAEs). Confirmation of biallelic pathogenic variants enables early consideration of emerging therapies like Otarmeni. Genetic counseling is advised for families to understand inheritance patterns, recurrence risks, and implications for future pregnancies.
Regulatory Pathway and Future Outlook
Otarmeni’s approval under the FDA’s National Priority Voucher (CNPV) pilot program reflects an evolving regulatory approach to incentivize development of treatments for rare and serious conditions. The CNPV program awards transferable vouchers for priority review to sponsors of approved products targeting designated rare diseases. These vouchers can be used to secure expedited FDA review for subsequent applications or sold to other companies.

As the first gene therapy approved under this program, Otarmeni sets a precedent for future nucleic acid-based therapies targeting sensory disorders. Researchers are exploring similar dual-vector strategies for other genes associated with hearing loss, such as GJB2 (connexin 26) and STRC (stereocilin), which together account for a significant proportion of genetic hearing loss cases.
Long-term follow-up of treated patients will be essential to assess the durability of transgene expression, potential immune responses to the AAV capsid, and the need for re-administration. Preclinical studies suggest that AAV vectors can persist in non-dividing cells like hair cells for extended periods, but clinical confirmation is awaited.
Regeneron has stated its commitment to working with healthcare providers, patient advocacy groups, and regulatory bodies to ensure equitable access to Otarmeni. Information about treatment centers, eligibility requirements, and clinical trial opportunities is expected to be made available through official channels, including the company’s website and the FDA’s approved drug products database.
As the medical community continues to evaluate the real-world impact of Otarmeni, the approval represents a milestone in the pursuit of precision medicine for sensory disorders. It validates the feasibility of gene therapy for inner ear diseases and opens the door to future innovations aimed at preserving and restoring hearing through genetic intervention.
For updates on Otarmeni, including prescribing information, safety updates, and access programs, patients and caregivers are encouraged to consult the FDA’s website or speak with a qualified healthcare provider specializing in genetic hearing disorders.
We welcome your thoughts and experiences. Share your comments below and help spread awareness by sharing this article with others who may benefit from this important development.