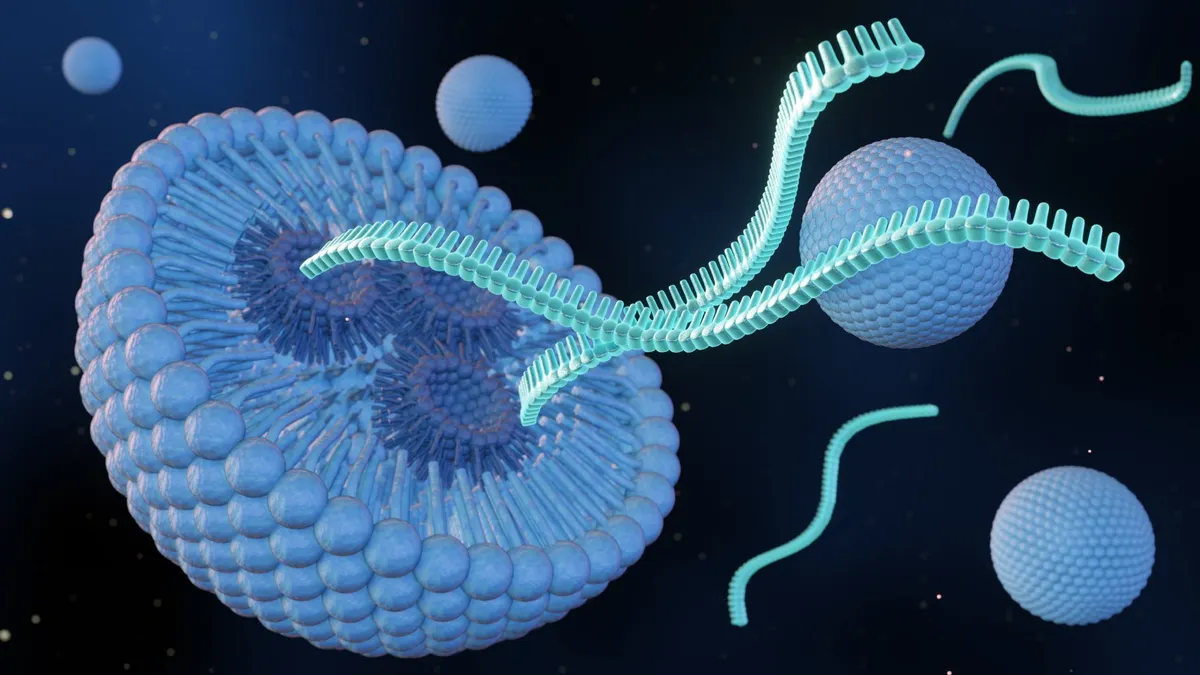
UCLA researchers have developed a lipid nanoparticle-based gene-editing approach that can insert an entire healthy gene into human airway cells, offering a potential new path toward mutation-agnostic therapy for cystic fibrosis. The method, described in a study published in Advanced Functional Materials on February 17, 2026, uses engineered lipid nanoparticles to deliver the molecular components needed for precise gene insertion without relying on viral vectors. This advancement addresses a key limitation of current treatments, which are ineffective for about 10% of cystic fibrosis patients who produce little or no functional CFTR protein.
The cystic fibrosis transmembrane conductance regulator (CFTR) gene, when mutated, leads to the buildup of thick mucus in the lungs, causing chronic infections and progressive lung damage. While CFTR modulator therapies have transformed care for many patients, they require some residual protein function to work. The UCLA approach aims to overcome this by delivering a full, healthy copy of the CFTR gene directly into airway cells, restoring chloride and water transport at the cellular level. In laboratory models, the treatment successfully restored key biological functions associated with normal airway cell activity.
Dr. Steven Jonas, senior author of the study and a member of the UCLA Broad Stem Cell Research Center, emphasized the significance of the non-viral delivery system. “This work shows that One can package everything needed for precise gene insertion into a single, non-viral delivery system,” he said. “That’s a critical step toward developing gene therapies that can work across many different disease-causing mutations.” The lipid nanoparticles used in the study are similar in structure to those employed in mRNA vaccines but were re-engineered to accommodate the larger genetic cargo required for full-gene insertion.
The research builds on years of investigation into nucleic acid delivery systems, with lipid nanoparticles emerging as a promising alternative to viral vectors due to their lower risk of immune response and insertional mutagenesis. By avoiding viral components, the approach may reduce safety concerns associated with long-term gene therapy while enabling repeat administration if needed. The study demonstrated successful gene insertion and expression in human bronchial epithelial cells cultured to mimic the airway environment.
Cystic fibrosis affects more than 70,000 people worldwide, with approximately 1,000 new cases diagnosed each year. Though modulator therapies like ivacaftor and elexacaftor-tezacaftor-ivacaftor have significantly improved lung function and quality of life for many, a subset of patients with nonsense or splicing mutations derive limited benefit. For these individuals, gene replacement strategies represent one of the few remaining avenues for potentially curative intervention. The UCLA team’s work contributes to a growing body of evidence supporting non-viral gene editing as a viable platform for treating monogenic lung diseases.
Beyond cystic fibrosis, the lipid nanoparticle platform could be adapted to address other inherited lung conditions caused by single-gene mutations, such as alpha-1 antitrypsin deficiency or surfactant protein deficiencies. The flexibility of the system—its ability to carry large DNA constructs alongside editing machinery—makes it suitable for a range of gene insertion, correction, or modulation strategies. Researchers note that further optimization is needed to improve delivery efficiency in vivo and to assess long-term safety and durability of gene expression in animal models before human trials can be considered.
The study was supported by funding from the National Institutes of Health and the Cystic Fibrosis Foundation, reflecting broader investment in genetic therapies for rare diseases. As of early 2026, several clinical trials are underway evaluating CRISPR-based and viral vector gene therapies for cystic fibrosis, but non-viral approaches like this one remain in preclinical development. Researchers at UCLA and collaborating institutions plan to next test the nanoparticle system in animal models to evaluate biodistribution, immune response and functional recovery in lung tissue.
While the laboratory results are promising, experts caution that translating gene-editing technologies from cell cultures to human lungs involves significant hurdles, including achieving sufficient transfection rates in vivo, overcoming mucosal barriers, and ensuring sustained expression without adverse effects. The field continues to balance innovation with caution, particularly given the history of immune reactions observed in early gene therapy trials. Any future clinical application would require rigorous preclinical toxicology studies and phased human testing under regulatory supervision.
For patients and families affected by cystic fibrosis, advances in gene editing represent a source of hope for treatments that go beyond symptom management to address the root genetic cause. Organizations such as the Cystic Fibrosis Foundation continue to fund research into genetic therapies and provide updates on ongoing studies through their official website and patient registries. As scientific understanding of delivery mechanisms improves, lipid nanoparticles may play an increasingly vital role in the next generation of genetic medicines.
The next step in this research path involves preclinical testing in animal models to assess safety, efficacy, and durability of gene expression. No human trials have been announced as of April 2026, and timelines for clinical translation remain uncertain pending further data. Researchers emphasize that while the approach shows promise, This proves one of several strategies being explored to expand treatment options for all individuals living with cystic fibrosis.
If you found this information helpful, please consider sharing it with others who may be interested in developments in genetic medicine. We welcome thoughtful comments and questions below—let us realize what aspects of this research you’d like to see explored in future coverage.
Worth a look